
Rett syndroom wordt veroorzaakt door een genetische mutatie, verandering van een gen, op het X-chromosoom. Rett syndroom is maar voor 1% erfelijk en het gaat dus vrijwel altijd een spontane mutatie. Sinds 1999 is bekend dat een mutatie van het MECp2 gen de oorzaak is van Rett syndroom. Binnen het MECP2 gen zijn er verschillende mutaties mogelijk op specifieke stukjes van het gen. Het MECP2 gen is een zogenaamd regulerend gen. Dit wil zeggen dat het bepalend is voor de activiteit van andere genen, die een rol spelen in de ontwikkeling van het centrale zenuwstelsel. Die andere genen maken eiwitten of enzymen aan die nodig zijn voor een normale ontwikkeling.
De symptomen van Rett syndroom treden op als gevolg van het achterblijven van de rijping van met name de hersenstam.
Rett syndroom bij jongens
Er wordt vrijwel altijd gesproken over Rett syndroom bij meisjes (vrouwen). Rett syndroom bij jongens is namelijk zeer zeldzaam. Er dient dan tevens sprake te zijn van het syndroom van Klinefelter, waarbij er i.p.v. één X en één Y chromosoom twee X- en één Y-chromosoom zijn (XXY). Ook kan er sprake zijn van een zgn. mosaïcisme, waarbij de lichaamscellen deels normaal zijn en deels gemuteerd MECP2 bevatten.
Meer uitleg oorzaak Rett syndroom
Het MECP2-gen ligt op de lange arm van het X-chromosoom (Xq28) en bestaat uit 4 exons. De coderende sequenties liggen in exon 2, exon 3 en exon 4. Het gen codeert voor het methyl-CpG-bindend proteïne 2, ook wel het MeCP2-eiwit genaamd. In meer dan 80% van de gevallen zijn MECP2-mutaties geïdentificeerd, tot 90 % in klassiek Rett syndroom en wat minder in de atypische gevallen. In ongeveer 20 % van de gevallen zijn (nog) geen MECP2-mutaties gevonden maar het is zeer onwaarschijnlijk dat de oorzaak van het Rett syndroom nog door een ander gen wordt bepaald. Vooral bij de atypische Rett meisjes en vrouwen verwacht men wel dat mutaties in andere genen dan MECP2 zullen worden gevonden.
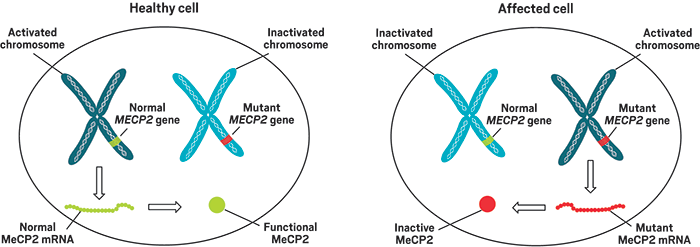
Het MeCP2-eiwit is een DNA-bindend proteïne dat in alle lichaamscellen aanwezig is maar in de hersenen in hoge concentraties voorkomt. Het is opgebouwd uit een methyl-CpG-bindend domein (MBD), een transcriptie-repressiedomein (TRD) en twee nucleaire lokalisatiesignalen (NLS), en eindigt met een terminaal C-segment. Het coderende deel van het MBD is verdeeld over exon 3 en exon 4, terwijl het coderende deel van het TRD geheel in exon 4 is gelegen. Het MeCP2-eiwit bindt met zijn MBD aan specifieke plaatsen binnen het DNA, terwijl het TRD ervoor zorgt dat het betreffende stuk en het omringende DNA niet meer toegankelijk zijn. De NLS zorgen voor het transport van MeCP2 in de nucleus.
Deze interacties resulteren in een repressie van transcriptie, die nodig is om de ruis van het omgevende DNA uit te schakelen en de targetgenen tot expressie te brengen. Vermoedelijk zorgen de mutaties voor een instabiel eiwit, dat sneller degradeert en minder lang zijn werking kan uitoefenen, waardoor het niet optimaal kan functioneren. Dit leidt tot onvoldoende onderdrukking en vervolgens tot over-expressie van een aantal genen, wat een potentieel beschadigend effect heeft op het zich ontwikkelende centrale zenuwstelsel.
Dit mechanisme stemt overeen met neuropathologische bevindingen die aantonen dat de hersenen het meest onderontwikkelde orgaan zijn bij meisjes met het Rett syndroom. De hersenen blijken in verhouding tot de lengte van de meisjes geringer te zijn in gewicht en omvang. Recent ontdekte men ook dat MeCP2-expressie significant hoger is in weefsel van het centraal zenuwstelsel dan in weefsel van het niet-centraal zenuwstelsel en dat MECP2-mutaties vermoedelijk alleen duidelijk worden in CZS-cellen die een zeer hoge MeCP2-expressie vertonen.
CDKL5, FOFG-1 en Mecp2 duplicatie syndroom
Recent zijn ook andere mutaties beschreven zoals CDKL5 en FOX-G1 die een beeld van Rett syndroom geven. Bij CDKL5 is het op jonge leeftijd optreden van epilepsie typerend. Het testen op CDKL5 mutatie is belangrijk bij een klinisch beeld van Rett zonder een afwijking in het MECP2 gen.
Sinds 2005 weten we dat het MECP2 gen betrokken is bij nog een ernstige neurologische ontwikkelingsstoornis: het MECP2 Duplicatie Syndroom. Bij dit syndroom is er geen mutatie op het MECP2 gen, maar is het gen in zijn geheel verdubbeld. In tegenstelling tot het Rett Syndroom komt deze aandoening juist met name voor bij jongens. Meisjes met de duplicatie zijn draagster en hebben over het algemeen geen symptomen.
Lees meer hierover op deze website.
Filmpje: https://youtu.be/eTzfA5_CdXg



